脊髓性肌萎缩症又称脊肌萎缩症(SMA),它是一种常见的罕见病,临床表现的症状为肌无力、肌萎缩,是一类由脊髓前角运动神经元变性导致肌无力、肌萎缩的常染色体隐性遗传病。

关注罕见病“脊髓性肌萎缩症”
什么是脊髓性肌萎缩症
脊髓性肌萎缩是一种遗传性疾病,其特征是用于运动的肌肉(骨骼肌)虚弱和萎缩,是由控制肌肉运动的运动神经元这种特殊神经细胞的缺失引起的,肌肉萎缩通常随着年龄的增长而加剧。
有多种不同类型的脊髓性肌萎缩是由相同基因的改变引起的,其常见症状如进行性肌痉挛等。
脊髓性肌萎缩症患者的生活
1、0型SMA: 在出生时就很明显,是最严重的一种SMA。致病的婴儿在子宫时就会出现胎动过少的现象,从而导致这些婴儿出生时往往伴随关节畸形、肌张力弱、呼吸衰竭(呼吸肌萎缩)甚至先天性心脏缺陷。这导致他们往往无法活过婴儿期。
2、I型SMA: 也称为威德森-霍夫曼综合征(werdnig- hoffmann syndrome),这是SMA的常见类型。这是一种严重的肌肉无力症,在出生时或出生后的头几个月就很明显,致病儿童无法控制他们的头部运动,也无法独自坐着。这种类型的儿童通常会有吞咽问题,导致喂养困难,从而出现发育不良。他们还会因为呼吸肌萎缩导致钟形胸和呼吸困难。大多数I型SMA的儿童由于呼吸衰竭而死亡。
3、II型SMA: 也称为杜博维茨病(Dubowitz disease),其特征是6-12个月时的肌无力,起初这种类型的儿童可以在没有支撑的情况下坐着,然而,随着肌肉无力不断恶化,这些儿童必须得到支撑才能坐着,如果没有支撑,甚至不能站立或行走。他们的手指经常会不由自主地颤抖,脊柱弯曲(脊柱侧弯),呼吸肌无力导致呼吸衰竭最终可能危及生命,II型SMA患者的寿命在20-30岁之间。
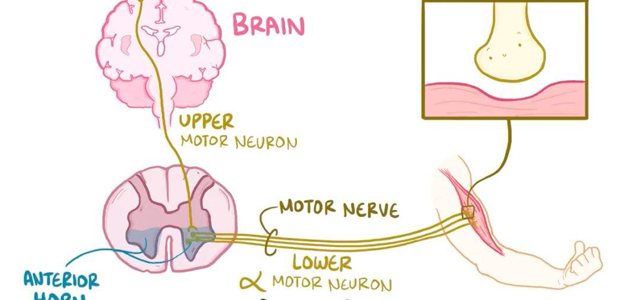
不同类型的脊髓性肌萎缩症危害不同
4、III型SMA: 也称为库格伯格-维兰德病( Kugelberg-Welander disease)通常在儿童早期就会引起肌肉无力,患有这种疾病的儿童可以独立站立和行走,但随着时间的推移,行走和站立会变得越来越困难,最终只能依靠轮椅。III型SMA患者由于没有呼吸肌无力,所以寿命与常人无异。
5、IV型SMA: 这类患者非常罕见,其肌无力现象通常在成年时才发生,患者通常会出现轻微到中度的肌无力和轻微的呼吸问题,一般认为,IV型SMA患者的预期寿命和正常人一样。
全世界每8,000到10,000人中就有1人患有SMA。I型SMA是最常见的类型,约占所有病例的一半,其次是II型和III型,而0型和IV型非常少见。
脊髓性肌萎缩症致病原因
SMN1基因突变导致上述所有类型SMA的发生,而SMN2基因的拷贝数变化,则决定了SMA的严重程度,所以一般通过检测SMN1和SMN2,就可以提前预知SMA的疾病类型。
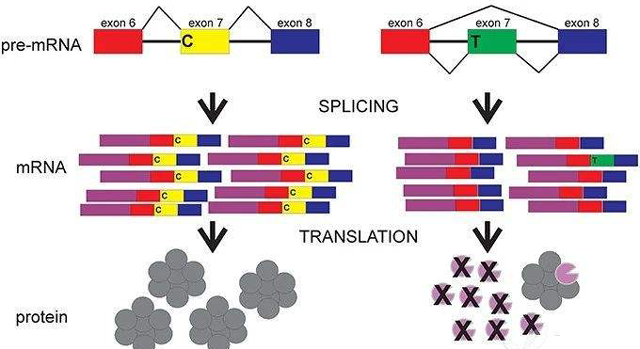
smn1与smn2基因表达示意图
SMN1和SMN2基因都提供制造一种叫做存活运动神经元(SMN)蛋白的指令。正常情况下,大多数功能性SMN蛋白由SMN1基因产生,少量由SMN2基因产生。SMN蛋白是一种被称为SMN复合体的蛋白质,它对运动神经元的维持非常重要。运动神经元传递来自大脑和脊髓的信号,告诉骨骼肌紧张(收缩),从而使身体运动。
患有SMA的患者都有SMN1基因缺失,而这会影响SMN蛋白的产生。缺乏SMN蛋白会导致信号无法在大脑和肌肉之间传递,最终造成运动神经元死亡。肌肉的收缩离不开大脑的信号,许多骨骼肌由于接受不到信号,只会逐步变弱、萎缩。
脊髓性肌萎缩症怎么遗传
SMA是一种常染色体隐性遗传,这意味着患者的父母都携带有突变的SMN1基因。父母通常不会变现出相关疾病症状,而他们的后代有25%的几率致病。
脊髓性肌萎缩症能治吗
FDA已经批准了两种治疗SMA的药物:nusinersen (Spinraza)和onasemnogene abeparvoveci -xioi (Zolgensma)。这两种药物都是典型的基因治疗药物,是通过调节SMN1和SMN2基因,使其产生更多的SMN蛋白,让身体能够提供传递大脑信号的指令,从而帮助患者控制肌肉运动。
治疗脊髓性肌萎缩症的药物
其中Nusinersen主要调节SMN2基因,适合全年龄的SMA患者使用,而Onasemnogene abeparvovec-xioi主要调节SMN1基因,适合2岁以下儿童使用。
脊髓性肌萎缩症生育干预
由于SMA是隐性的遗传病,有些人表面上是正常的,其实是该基因病的携带者,那么该如何知道是否携带呢?目前只能通过基因检测来完成,如果无法确认自己是否携带有SMN1和SMN2基因异常。
如果父母是SMA的携带者,又该如何阻断SMA的遗传?由于SMA是隐性的遗传病,可以通过三代试管婴儿技术筛查健康的胚胎,从而阻断该病的遗传,目前国际先进分子诊断技术,可以阻断已知所有的单基因疾病包括SMA。
遗传疾病大全