面肩肱型肌营养不良症是以肌无力和消瘦(肌肉萎缩)为特征的疾病。这种情况得名于最常受影响的肌肉:脸部,肩胛骨和上臂的肌肉。面肩肱型肌营养不良症的体征和症状通常出现在青春期。但是,病情的发生和严重程度差别很大。较温和的病例可能在晚年才会变得明显,而罕见的严重病例在婴儿期或童年早期就显现出来。
面部肌肉或肩膀的虚弱通常是这种情况的首要症状。面部肌肉无力使得难以用习惯喝水和吹口哨,或微笑时转动嘴角。眼睛的肌无力导致在睡着时眼睛无法完全闭合,从而导致眼睛干涩等眼部问题。脸部一侧的软弱可能比另一侧更为严重,由于不清楚的原因。肩部肌肉欠佳倾向于使肩胛从背部突出,这是一种常见的肩胛骨翼状体征。肩膀和上臂的肌肉虚弱可能会使抬起手臂或掷球困难。
面肩肱型肌营养不良症
与面肩肱型肌营养不良相关的肌无力在数十年内缓慢恶化,并可能扩散到身体的其他部位。小腿肌肉的虚弱可能会导致一种称为足部下垂的情况,这会影响步行并增加跌倒的风险。臀部和骨盆肌肉无力可能使爬楼梯或走路很困难。由于腹部肌肉较弱,受影响的个人可能会有一个夸张的腰部弯曲(脊柱前凸)。大约20%的受影响个人最终需要使用轮椅。
其他体征和症状面肩肱型肌营养不良症可以包括轻度高音听力损失和在视网膜的光敏感组织异常。这些迹象往往不明显,只能在医疗检测过程中发现。很少,面肩肱型肌营养不良会影响呼吸所需的心脏肌肉或肌肉。

20%面肩肱型肌营养不良症患者需要坐轮椅
研究人员描述了两种类型的面肩肱型肌营养不良症:1型(FSHD1)和2型(FSHD2)。这两种类型具有相同的体征和症状,并以其遗传原因来区分。
面肩肱型肌营养不良症发病率
面肩肱型肌营养不良症估计发病率为1万人。所有病例中大约95%是FSHD1; 剩下的5%是FSHD2。
面肩肱型肌营养不良症的发病原因
面肩肱型肌营养不良症是由涉及4号染色体长臂(q)的遗传变化引起的。这种疾病的两种类型都是由染色体末端附近的DNA区域(称为D4Z4)的变化引起的。该区域由11到100多个重复片段组成,每个片段长约3300个DNA碱基对(3.3kb)。整个D4Z4区域通常是超甲基化的,这意味着它具有大量与DNA相连的甲基(由一个碳原子和三个氢原子组成)。甲基使基因沉默,因此DNA的高甲基化区域倾向于具有更少的打开(有效)的基因。面肩肱型肌营养不良症结果当D4Z4区域低甲基化,缺少连接的甲基。在FSHD1中,发生低甲基化是因为D4Z4区域异常缩短(收缩),包含1到10次重复,而不是通常的11到100次重复。在FSHD2中,低甲基化通常是由称为SMCHD1的基因中的突变引起的,其提供了制造通常高甲基化D4Z4区域的蛋白质的说明。然而,大约20%的FSHD2患者在SMCHD1基因中没有确定的突变,并且低甲基化的原因是未知的。

面肩肱型肌营养不良症会导致面部肌肉无力
D4Z4区域的高甲基化通常在大多数成人细胞和组织中保持称为DUX4的基因沉默。所述DUX4基因位于最靠近的端部的D4Z4区的区段4号染色体。在面肩肱型肌营养不良症患者中,D4Z4区低甲基化可防止DUX4基因在通常被关闭的细胞和组织中沉默。尽管对DUX4基因活性的功能知之甚少,但研究人员认为它影响其他基因的活性,特别是在肌细胞中。DUX4的异常活动是未知的基因破坏或破坏这些细胞,导致进行性肌无力和萎缩。
所述DUX4基因毗邻的DNA的调节区上的4号染色体被称为PLAM序列,这是必要的生产DUX4蛋白质。4号染色体的一些拷贝具有功能性pLAM序列,而另一些拷贝不具有。具有功能性pLAM序列的4号染色体的拷贝被描述为4qA或“允许的”。没有功能性pLAM序列的那些被描述为4qB或“不允许的”。没有功能性pLAM序列,则不能制备DUX4蛋白。由于每个细胞中有4个4号染色体拷贝,所以个体可能有4个染色体的4个 “允许”拷贝,2个“不允许”拷贝或者每个拷贝中的一个。面肩肱型肌营养不良症只能发生在至少有一个“宽容” 4号染色体拷贝的人身上。无论受影响的个体是否具有感染的D4Z4区域或SMCHD1基因突变,仅当功能性pLAM序列也存在以允许DUX4蛋白产生时才会导致疾病。
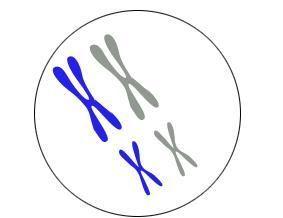
面肩肱型肌营养不良症由4号染色体异常引起的
研究表明,导致FSHD2 的SMCHD1基因突变也可能增加FSHD1患者的疾病严重程度。研究人员怀疑,合并D4Z4区域和SMCHD1基因突变的组合导致D4Z4区域连接的甲基更少,这使得DUX4基因具有高度活性。在两种基因改变的人中,过度活跃的基因导致严重的肌无力和萎缩。
面肩肱型肌营养不良症的遗传模式
FSHD1是以常染色体显性模式遗传的,这意味着在“允许” 染色体4上的缩短的D4Z4区域的一个拷贝足以引起疾病。在大多数情况下,受影响的人从一个受影响的父母遗传改变的染色体。其他FSHD1患者没有家族史。这些情况被描述为零星的,是由一个新的(从头)D4Z4在“宽容” 染色体4的一个拷贝上收缩。
FSHD2是以双基因模式遗传的,这意味着两个独立的遗传变化是引起疾病的必要条件。对于FSHD2,个体必须继承SMCHD1基因(其位于第18号染色体上)的突变,并且分别地,它们必须继承“允许” 染色体4的一个拷贝。受影响的个体通常继承一个亲本的SMCHD1基因突变和来自另一个亲本的“允许” 染色体4。(因为在大多数情况下,父母都没有遗传改变,所以他们通常不受影响。)