马凡氏综合症是什么
马凡氏综合征又叫马凡综合征、先天性中胚层发育不良,它是一种比较少见的先天遗传性结缔组织疾病,造成的原因为常染色体显性遗传,伴随有家族史。
| 病症名称 | 马凡氏综合征 | 英文名称 | Marfan syndrome |
| 别称 | 马凡综合征、先天性中胚层发育不良、蜘蛛指征、肢体细长症 | ||
| 就诊科室 | 心血管外科 | 类型 | 遗传性结缔组织疾病 |
| 致病原因 | 常染色体显性遗传,原纤维蛋白-1基因是最常见致病基因 | ||
| 临床症状 | 身材高大、蜘蛛指、先天性心血管畸形、晶状体异位 | ||
| 患病明星 | 霍萱、朱刚、韩朋山、张佳迪、武强等 | ||
虽然马凡氏综合征能够带给患者异于常人的运动体格,但患者往往伴随有心血管系统异常,特别是合并的心脏瓣膜异常和主动脉瘤,不注意情况下会导致猝死,可谓体内的“不定时炸弹”。

马凡综合征手指、脚趾细长不匀称
患有马凡氏综合症的患者常表现的症状为体格细高、四肢及指(趾)细长,特别是四肢、手指、脚趾细长不匀称。95%的马方综合征患者死于心血管并发症,常见为动脉瘤破裂和充血性心力衰竭。未经治愈的患者平均寿命为男性30岁左右,女性40岁左右。
马凡氏综合征的发病率约为0.04‰~0.1‰,据推测中国大约有16万人患有此病。要知道马方综合征虽然不能根治,但越早发现对于后期预后有着很大的作用,同时也能延长患者生命。
马凡氏综合症的症状有哪些
马凡氏综合症是由于人类第15号染色体上的原纤维蛋白基因(Fibrillin-1,FBN1)缺陷所致。原纤维蛋白单体是由42个氨基酸所组成的多肽,是弹性纤维的基本成分,它也完全依赖于钙和半胱氨酸二硫键的结合。
身材高大、蜘蛛指是马凡氏综合症的常见症状
当马凡氏综合症时,半胱氨酸被其它氨基酸所替代,不能与钙结合,导致弹性纤维变性、细小和退变,从而引起动脉层中的囊性坏死和断裂,病理上可发现III-IV级中层退行性变。
关于马凡综合征的症状常见的表现为患者多体型颀长,柔软度超人。有数据显示在儿童时期马凡氏综合征患者39%无心血管病变,仅有身高增长速度过快而体重增长相对迟缓。
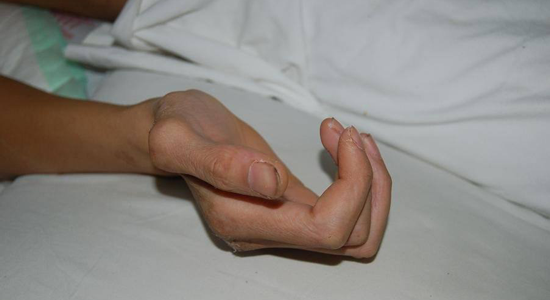
马凡氏综合症患者的手异于常人
儿童首次诊断为马凡氏综合症大多是根据心血管以外的一些症状表现,比如75%是近视(晶状体半脱位),83%是脊检侧弯或者蜘蛛指(趾)等其他畸形。
成年马凡氏综合征患者的话,除了体格异常与关节松弛等症状外,主要是心脏功能不全及由于升主动脉瘤压迫出现的症状,如心悸、胸闷、气短、胸痛等。
“天才病”马凡氏综合征的诊断方法
马凡氏综合征的诊断标准最早在1987年第七届人类遗传学国际大会上建立,并于1988年第一次国际马凡氏综合征专题讨论上明确规定,随后1996年de Paepe A重新修正被视为统一标准。
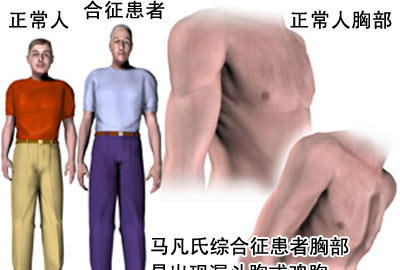
马凡氏综合症患者胸部易出现漏斗胸或鸡胸
骨骼系统
针对骨骼系统的诊断标准分为主要和次要,是否为马凡综合征患者,需符合的条件包括至少两项主要标准或一项主要标准加两项次要标准。
骨骼系统症状:
- 鸡胸;
- 漏斗胸需外科矫治;
- 上部量/下部量的比例减少,或上肢跨长/身高的比值大于 1.05;
- 腕征、指征阳性;
- 脊柱侧弯大于 20 度,或脊柱前移(侧弯计);
- 肘关节外展减小(〈170度);
- 中踝中部关节脱位形成平足;
- 任何程度的,髋臼前凸(髂关节内陷)(X 片上确定)。
次要标准:中等程度的漏斗胸:关节活动异常增强;高腭弓,牙齿拥挤重叠;面部表征:长头——正常头颅指数为 75.9 或以下、颧骨发育不全、眼球内陷、缩颌、睑裂下斜。
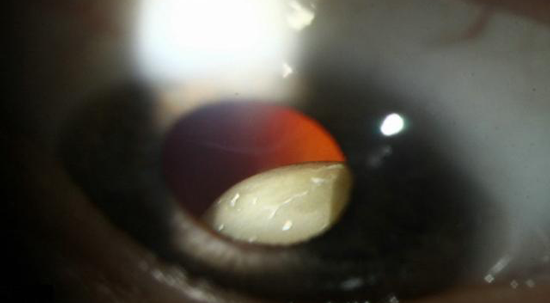
针对眼睛的判断标准:晶状体脱位
眼睛系统
主要标准是晶状体脱位,次要标准包括异常扁平角膜(角膜曲面计测量);眼球轴长增加(超声测量);虹膜或睫状肌发育不全致瞳孔缩小。眼睛系统受累需符合标准:主要标准或至少两项次要标准。
心血管系统
心血管受累需符合的条件:有一项主要标准或一项次要标准即可。
次要标准
- 二尖瓣脱垂伴或不伴二尖瓣返流;
- 主肺动脉扩张( 在无瓣膜或外周肺动脉狭窄及其它明显原因下,年龄又小于 40 岁);
- 二尖瓣环钙化(年龄小于 40 岁);
- 降主动脉或腹主动脉扩张或夹层(50 岁以下)。
主要标准:升主动脉扩张伴或不伴主动脉瓣返流,以及至少 Valsava 氏窦扩张;升主动脉夹层。
其他
其他包括肺系统、皮肤和体包膜、家族或遗传史等,具体如下:
肺系统
- 自发性气胸;肺尖肺大泡(胸片证实)。
肺系统
皮肤和体包膜
- 皮纹萎缩(牵拉痕),与明显超重、妊娠或反复受压等无关;复发性疝或切口疝。一项次要标准存在即可认为皮肤或体包膜受累。(次要标准)
皮肤和体包膜
家族或遗传史
- 父母、子女或兄弟姊妹之一符合该诊断标准;FBNI 基因中存在已知的导致马凡综合征的突变;存在已知的与其家族中马凡综合征患者相同的 FBNI 基因单倍型。(主要标准)
家族或遗传史
注:由于家族或遗传史在诊断中意义重大,主要标准中必须有一项存在。
马凡综合征的诊断前提
对特定病例:如果无家族或遗传史者,至少需有两个不同系统的主要标准以及三分之一的器官受累;如果检出一个已知马凡综合征的基因突变,一个系统中有一项主要标准和第二项系统受累即可诊断。
需与马凡综合征鉴别的疾病
- 先天性挛缩性蜘蛛指征(Viljoen,et al.1994);
- 家族性胸主动脉瘤(Savunen 1987;Emanuel,et al. 1977), 过去称为埃德海姆囊性中层坏死;
- 家族性主动脉夹层(Nicod,et al. 1989);
- 家族性晶状体脱位(Tsipouras,et al.1992);
- 家族性类马凡体型(Milewicz,et al.1995);
- 家族性二尖瓣脱垂综合征(Devereux,et al.1982,1986,1987 ; Roman ,1989b);
- Shprintzen-Goldberg 综合征(Shprintzen和 Goldberg,1982);
需与马凡综合征鉴别的疾病
对特定病例的家属:在家族史中有一项主要标准、一个系统的一项主要标准和第二个系统受累即可诊断。
关于马凡氏综合症新诊断标准与1988年的诊断标准的主要区别在于,对家族中无典型马凡综合征患者的诊断要求更为严格,同时骨骼作为一项主要标准,8 项表现中必须至少有4项方可考虑骨骼受累等。
马凡氏综合症患者真人照片
因马凡氏综合征发生的悲剧不少,早期中国女排前国手霍萱、美国女排名将海曼、四川男排国手朱刚以及男篮运动员张佳迪和武强均因此病猝死,“马凡氏”已夺走多名运动员生命,以下是部分马方综合征患者真人照片:

蜘蛛人”常继国
钱峰伟的手指细长,这是马凡氏综合征患者最具代表性的蜘蛛指
马凡氏综合征除极少数是基因突变而来外,大部分是遗传产生的。病人多患有心血管系统异常,一旦出现主动脉异常随时会危及生命,如果不及时手术,存活的希望几乎为零。
马方综合征治疗方法
其实现实中针对马凡氏综合征的诊断并非难事,一般经过检查和询问病史、家族史,并经彩色B超和磁共振检查后即可确诊。而一旦确诊的话就要根据情况进行手术治疗,只有早发现早治疗,才能避免心力衰竭、主动脉破裂和心肌梗塞的发生。
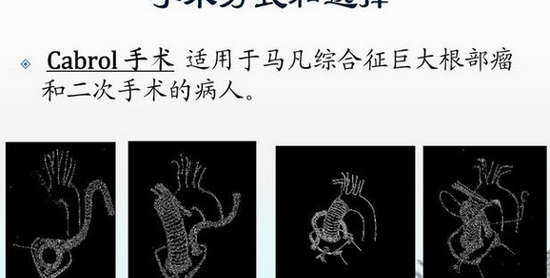
手术治疗会根据患者情况进行选择
一般性药物治疗
针对马凡氏综合征的治疗方法一般建议手术为主,因为目前来说药物治疗或其他保守疗法均不能去除此病。药物治疗的主要目的是减缓或推迟心血管病变的发生,防止心率失常。
手术治疗
随着科技进步,目前马凡氏综合征手术成功率已在90%以上。当马凡氏综合征有出现心血管病变时就只能通过手术治疗。一旦出现心血管病变,就必须马上治疗,否则预后极差。
手术治疗适应症
- 主动脉根部径>50mm;
- 有家族史(猝死或主动脉夹层)主动脉根部径≥45mm;
- 主动脉根部增长率>5mm/年;
- 儿童根部瘤直径扩张大于其正常直径50%应手术治疗。
可能有人会担心马凡氏综合征的治疗费用昂贵负担不起,实际上马凡氏综合征治疗费用与做主动脉瘤的手术费用差不多,都是在15万左右。如果是发现早、治疗早,病变没有出现主动脉夹层,或者说没有广泛病变,这种情况下治疗彻底且费用较低。
相关疑问
Q:马凡氏综合征是否为禁止结婚的遗传疾病?
A:不是,马凡氏综合征是常染色体显性遗传,如果夫妻俩决定要孩子的话建议去做基因检测的机构查一下染色体,如有染色体异常那么可通过第三代试管婴儿进行筛查做到优生优育。