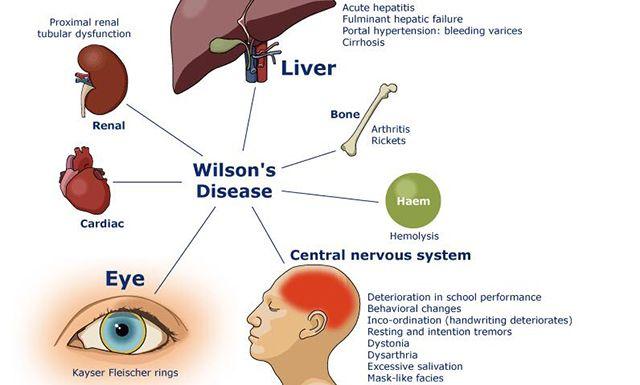
肝豆状核变性(Wilson’s disease, WD),常染色体隐性遗传性疾病,青少年多见,是先天性铜代谢障碍性疾病。由Wilson首先报道和描述,是一种遗传性铜代谢障碍所致的肝硬化和以基底节为主的脑部变性疾病。临床上表现为进行性加重的锥体外系症状、肝硬化、精神症状、肾功能损害及角膜K-F环。WD对于青少年期起病,发病年龄4~50岁,以肝脏症状起病者平均11岁,以神经症状起病者平均19岁,起病多较缓慢。早期(尤其是症状前)诊断和及时、确切的治疗常可获得与健康人一样的生活和寿命。
肝豆状核变性
肝豆状核变性相关基因介绍
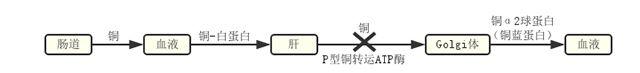
WD的致病基因为ATP7B,定位于13q14.3区,P型铜转运ATP酶,点突变为主。已发现WD其致病基因突变400余种,突变位点几乎遍布整个基因编码区,中国人群热点为p.Arg778Leu。突变导致铜代谢障碍,铜蓝蛋白减少,过多的铜沉积在肝、脑、肾、角膜等。
肝豆状核变性基因介绍
肝豆状核变性三代试管婴儿案例分享
基本信息
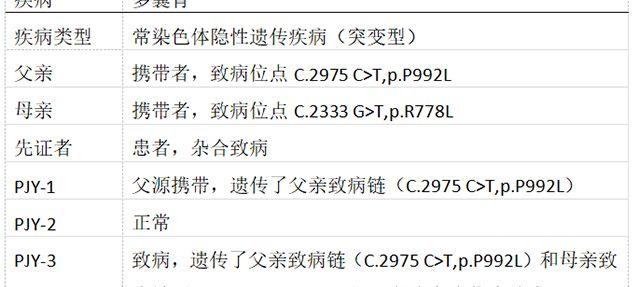
家系特点:此家系父亲致病位点(C.2975 C>T,p.P992L)、母亲致病位点(C.2333 G>T,p.R778L),先证者为遗传了父亲致病位点母亲致病位点的复合杂合位点致病。
父母均为WD携带者,女性先证者,申请PGD助孕。检测父母双亲、先证者、胚胎3个(PJY-1、PJY-2、PJY-3)。
肝豆状核变性检测方法
三代试管婴儿S-PGD——三重防护

肝豆状核变性做三代试管婴儿避免遗传
JBRH解决方案检测特点
- 检出率高,适用范围广;
- 检测结果准确,能够有效检测到重组,可无先证者;
- 靶向捕获设计,方案灵活,便于临床成本控制。
肝豆状核变性S-PGD检测结果
肝豆状核变性诊断结果
肝豆状核变性诊断结果2
妊娠结果:
经过PGS后,发现正常的PJY-2胚胎染色体异常,PJY-1胚胎染色体正常,最终移植PJY-1胚胎,已成功妊娠。
JBRH基于SNP连锁分析的S-PGD解决方案优势

肝豆状核变性做三代优势大
肝豆状核变性做三代试管婴儿临床意义:
- 更适用于临床使用:对每种疾病精细化设计,检测致病位点,可检测到重组,检测准确度更高;
- 分析结果更灵敏:测序不但能够利用设计好的SNP,还能发现新的SNP并加以利用,提高了重组断点的检测灵敏度;
- 帮助减少先天性无虹膜症患儿的出生,有利于优生优育。
遗传疾病大全