先天性无虹膜(congenital anirdia,AN)是一种先天性遗传性虹膜发育不良性疾病,是一种少见的眼组织畸形,于1819年由Barrata首先报道。目前认为,其发病率大致为1/10000~1/50000。AN常有家族史,有多达连续四、五代发病的家系报道,提示为常染色体显性遗传,外显率较高(84%)。国内有些报道显示:27个家系103例,患者子代132人中72人发病(54.6%),外显率为100%。目前尚无治愈先天性无虹膜疾病的有效方法,所以,遗传咨询和产前诊断对于预防无虹膜症的形成至关重要。
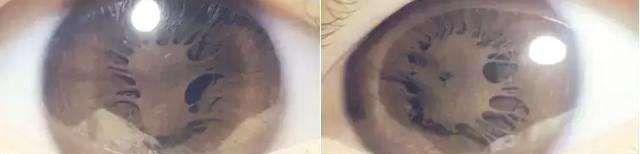
先天性无虹膜为常染色体显性遗传
先天性无虹膜有哪些症状表现
该病具有复杂的表型异质性,除虹膜组织缺如或缺损外,常伴有其他眼组织的结构异常,累及角膜、小梁网、晶状体、玻璃体和视网膜等,因此,很多患者往往合并角膜浑浊、青光眼、白内障、玻璃体浑浊、视网膜病变等眼部疾病。由于病变的轻重程度不同以及受累部位不同,不同患者个体间的视功能状态变异很大,即使在同一个家系的患者之间也可以出现不同的临床表现。
本病通常为双侧性,肉眼检查几乎看不到虹膜组织,但实际上并非完全没有虹膜。通过前房角镜检查可看到在前房角深部的虹膜残根。由于虹膜缺如,在大部分患者眼内可直接看到晶状体边缘与悬韧带。本病常伴有黄斑发育不良,所以患者视力往往很差,通常无法矫正,并有眼球震颤和畏光症状。
先天性无虹膜疾病相关基因介绍
AN遗传方式主要为:常染色体显性遗传,PAX6基因是AN的主要致病基因。PAX6基因是一种同源盒基因,位于染色体11p13上,有17个外显子,其mRNA大小为2341bp,编码含422个氨基酸的转录调节蛋白。该转录因子通过DNA结合域识别其他靶基因,通过转录激活域激活下游基因的表达。PAX6基因在眼、鼻、胰腺和中怄神经系统的发育中都起着重要作用,不同功能域出现的突变可有不同的临床表现。
先天性无虹膜三代试管婴儿案例分享
基本信息
家系特点:父亲为先天性无虹膜患者,PAX6基因内含子6存在c.357+5G>A杂合突变;母亲正常;先证者为PGD男方的母亲,同样为先天性无虹膜患者,存在c.357+5G>A杂合突变,寻求PGD助孕。
检测父母双亲、先证者1例,胚胎4个。
先天性无虹膜检测方法:
三代试管婴儿PGD——三重防护

三代试管婴儿PGD三重优势
JBRH解决方案检测特点
- 检出率高,适用范围广;
- 检测结果准确,能够有效检测到重组,可无先证者;
- 靶向捕获设计,方案灵活,便于临床成本控制。
先天性无虹膜的S-PGD检测结果
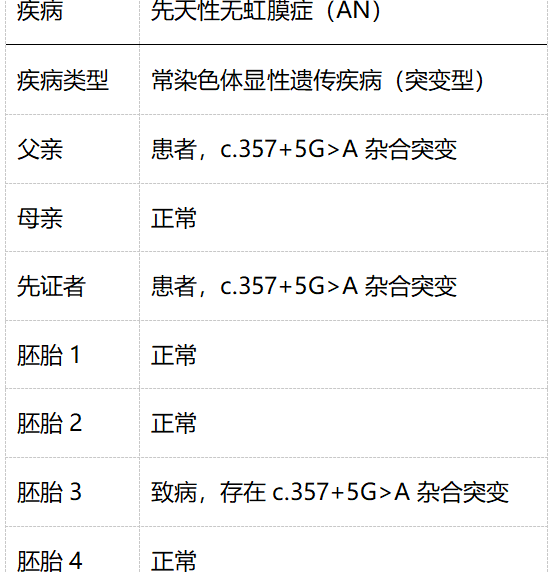
先天性无虹膜诊断结果

先天性无虹膜诊断结果
JBRH基于SNP连锁分析的S-PGD解决方案优势

三代试管婴儿解决先天性无虹膜的优势
先天性无虹膜采用三代试管婴儿的临床意义:
- 更适用于临床使用:对每种疾病精细化设计,检测致病位点,可检测到重组,检测准确度更高;
- 分析结果更灵敏:测序不但能够利用设计好的SNP,还能发现新的SNP并加以利用,提高了重组断点的检测灵敏度;
- 帮助减少先天性无虹膜症患儿的出生,有利于优生优育。
遗传疾病大全