先天性肌无力综合征是一组以肌无力(肌无力)为特征的疾病,随着体力消耗而加重。这种肌肉无力通常始于儿童早期,但也可能出现在青春期或成年期。面部肌肉,包括控制眼睑的肌肉,移动眼睛的肌肉,以及咀嚼和吞咽的肌肉,是最常受影响的。然而,任何用于运动的肌肉(骨骼肌)都可能在这种情况下受到影响。由于肌肉无力,受影响的婴儿可能有进食困难。诸如爬行或行走等运动技能的发展可能会延迟。肌无力的严重程度差别很大,一些人有轻微的虚弱,另一些人有严重的虚弱以至于不能行走。

先天性肌无力综合征患者袁好男高考640分
有些人会出现呼吸问题,可能是发烧或感染引起的。严重受影响的人还可能出现呼吸暂停(呼吸暂停),导致皮肤或嘴唇发青(发绀)。
先天性肌无力综合征的病因是哪些?
很多基因可以导致CMS,由于CMS是由于负责神经肌肉信号传递的蛋白质功能异常而致,根据突变蛋白的类型和位置,可分为四种类型:编码突触前膜蛋白质的基因突变(如ChAT)、编码突触间隙蛋白质的基因突变(如ColQ)、编码突触后膜蛋白质的基因突变(如AChR缺乏)及蛋白质翻译后糖基化缺陷和某些与疾病重叠的综合征(如GFPT1)。
这些基因发生突变导致在神经肌肉连接功能中起作用的蛋白质发生变化,并破坏神经细胞和肌肉细胞之间的信号传递。这些细胞间信号的中断会导致骨骼肌运动能力受损、肌肉无力和运动技能发育迟缓。而CMS引起的呼吸问题是由胸壁肌肉和分隔腹腔与胸腔(隔膜)的肌肉的运动受损引起的。
先天性肌无力综合征的遗传模式
多数CMS的遗传方式是常染色体隐性遗传,这意味着父母双方各自携带有一份突变基因,但他们通常不表现出这种情况的迹象和症状,而他们的后代则有25%的几率致病。
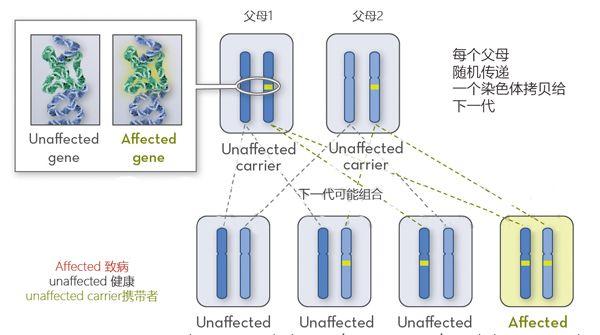
先天性肌无力常染色体隐性遗传概率
少数情况下,如SNAP25、synaptotagmin 2和慢通道CMS,是通过常染色体显性遗传的,这意味着父或母有一方患有该病,其后代有50%的几率致病。
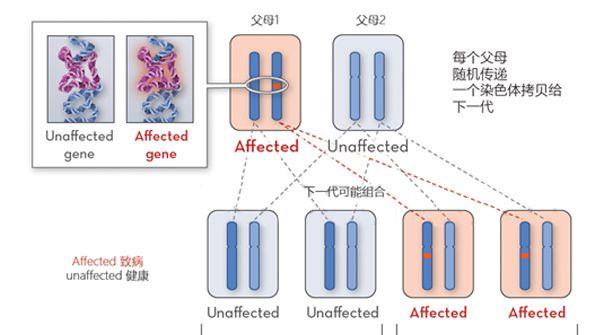
先天性肌无力常染色体显性遗传概率
先天性肌无力综合征要怎么治疗
由于CMS的致病基因不同,所以目前还没有统一的治疗标准,因为证明对一种CMS有效的药物对另一种CMS可能无效甚至有害,一般都是根据各个医生的临床经验和患者情况制定个性化的用药方案。
目前治疗CMS的药物包括胆碱能激动剂,如吡啶吡啶或氨法普丁(3,4-二氨基吡啶),长效开放通道阻滞剂乙酰胆碱受体离子通道氟西汀和奎尼丁,以及肾上腺素能激动剂,如沙丁胺醇和麻黄碱。
- AChR缺乏型CMSCHRNA1, CHRNB1, CHRND, CHRNE吡啶吡啶,氨法普丁,沙丁胺醇 /麻黄碱
- RAPSYN 型CMSRAPSYN吡啶吡啶,麻黄碱
- DOK-7 型CMSDOK-7吡啶吡啶,氨法普丁
- ChAT型 CMSChAT吡啶吡啶,沙丁胺醇
- 快通道型CMSCHRNA1, CHRNB1, CHRND, CHRNE吡啶吡啶,氨法普丁,沙丁胺醇 /麻黄碱
- 慢通道型CMSCHRNA1, CHRNB1, CHRND, CHRNE奎宁,氟西汀
先天性肌无力综合征的预后
先天性肌无力综合征发病率较低,其预后与突变的基因类型有关。病情进展较快的类型预后较差,可能在数年内致残甚至致命,而发展缓慢的类型预后相对较好。
先天性肌无力综合征怎么避免遗传
本病属于遗传病,有家族史者,应进行遗传咨询。由于该病的很多情况属于隐性的,可以通过携带者筛查,看是否是该遗传病的携带者,一方面可以通过携带者筛查,并可以通过三代试管婴儿技术筛查健康的胚胎,从而阻断该病的遗传,泰国试管婴儿医院同步国际先进分子诊断技术,可以阻断已知所有类型的CMS疾病。
遗传疾病大全